
.
.
 90
90
R. L. Mackman, J. Micklefield, M. H. Block, F. J. Leeper and A. R. Battersby
"Haem d1: Development of a new coupling procedure leading to the synthesis of isobacteriochlorins"
J. Chem. Soc., Perkin Trans. 1, 1997, 2111-2122.
Full Text
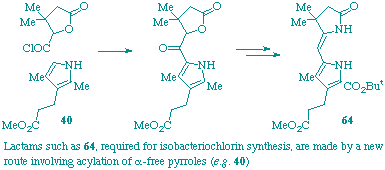
A new approach has been developed for construction of the western and eastern lactams needed for synthesis of isobacteriochlorins. It involves acylation of pyrroles with lactonic acids to form ketones. These are then efficiently converted into the desired lactams by a short sequence of reactions. All the steps are high yielding and simple to carry out.
.
.
.
.
.
.
.
 91
91
J. Micklefield, M. Beckmann, R. L. Mackman, M. H. Block, F. J. Leeper and A. R. Battersby
"Haem d1: Stereoselective Synthesis of the Macrocycle to Establish its Absolute Configuration as 2R,7R"
J. Chem. Soc., Perkin Trans. 1,1997, 2123-2138.
Full Text
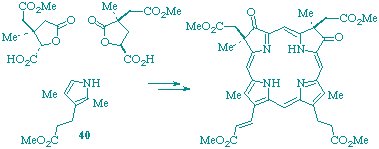
Although the gross structure of haem d1 has been established, the absolute stereochemistry at C-2 and C-7 is unknown. An unambiguous stereoselective synthesis of the ester of the metal-free macrocycle corresponding to haem d1 has been completed which establishes the absolute configuration of the natural cofactor as 2R,7R. Haem d1 is thus shown to match stereochemically other biologically important macrocycles, e.g. those involved in the biosynthesis of vitamin B12, which are related to isobacteriochlorins and also display 2R,7R configurations. The synthetic sequence used is based on a new procedure for assembly of the western and eastern building blocks and it serves as an efficient general route for construction of isobacteriochlorins.
.
.
.
.
.
.
.
.
.
.
 92
92
D. Appleton, A. B. Duguid, S.-K. Lee, Y.-J. Ha, H.-J. Ha and F. J. Leeper
"Synthesis of Analogues of 5-Aminolaevulinic Acid and Inhibition of 5-Aminolaevulinic Acid Dehydratase"
J. Chem. Soc., Perkin Trans. 1,1998, 89-101.
Full Text
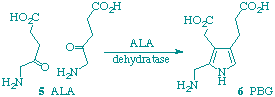
Syntheses are described of several analogues of 5-aminolaevulinic acid (ALA), which are potential inhibitors of ALA dehydratase (porphobilinogen synthase), an early enzyme of tetrapyrrole biosynthesis. Most of the analogues are relatively weak competitive inhibitors of the enzyme from Bacillus subtilis or irreversible inhibitors due to multiple alkylation of the enzyme but the 3-oxa and 3-thia analogues are potent mechanism-based inhibitors which inactivate, by acylation of a nucleophilic residue, probably the active-site lysine residue. The kinetics of the inactivation by 3-thiaALA have implications for the mechanism of the enzymic reaction.
.
.
.
.
.
.
.
93 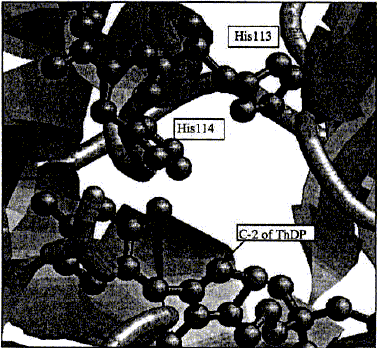 G. Schenk, F. J. Leeper, R. England, P. F. Nixon and R. G. Duggleby
G. Schenk, F. J. Leeper, R. England, P. F. Nixon and R. G. Duggleby
"The role of His113 and His114 in pyruvate decarboxylase from Zymomonas mobilis"
Eur. J. Biochem.,1997, 248, 63-71.
Full Text
Pyruvate decarboxylase (PDC) is one of several enzymes that require thiamin diphosphate (ThDP) and a divalent cation as essential cofactors. Recently, the three-dimensional structures of the enzyme from two yeasts have been determined. While these structures shed light on the binding of the cofactors and the reaction mechanism, the interactions between the substrate pyruvate and the enzyme remain unclear. We have used PDC from Zymomonas mobilis as a model for these enzymes in order to study substrate binding. The recombinant enzyme was expressed in Escherichia coli. High yield, simplicity of purification, high stability and simple kinetics make this model well suited for these studies. Activity measurements in the pH range between 5.8 and 8.5 indicated that a His residue may be involved in substrate binding. Analysis of an alignment of all known PDC protein sequences showed two invariant His residues (His113 and His114) which, according to the crystal structure of yeast PDC, are in the vicinity of the active site. Here we demonstrate that replacement of His114 by Gin does not have a great effect on cofactor and substrate binding. However, the kcat is decreased indicating that His114, may assist in catalysis. In contrast, substitution of His113 by Gin renders the enzyme completely inactive. This mutant has decreased affinity for both cofactors, as revealed by measurements of tryptophan fluorescence quenching. However, this decreased affinity is insufficient to account for the complete loss of activity. Despite its inability to support overall catalysis, this [Gln113]PDC mutant is capable of releasing acetaldehyde from 2-(1-hydroxyethyl)thiamin diphosphate supplied exogenously. It is proposed that upon substrate binding, His113 is placed close to C-2 of the thiazole ring. Subsequent deprotonation of this atom leads to a conformational change that allows a flexible loop (residues 105-112) that precedes His113 to close over the active site. Hence, replacement of His113 by another residue interferes with this closure of the active site and thus disrupts the catalytic process.
.
.
.
.
.
.
.
.
94 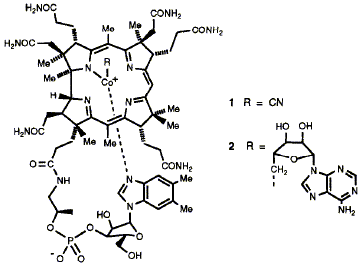 A. R. Battersby and F. J. Leeper
A. R. Battersby and F. J. Leeper
"Biosynthesis of Vitamin B12"
in Biosynthesis: Polyketides and Vitamins, Topics in Current Chemistry,Vol. 195, Springer-Verlag, Heidelberg, 1998, 143-193.
Full Text
Vitamin B12 (cyanocobalamin) is the normal isolated form of coenzyme B12 (adenosylcobalamin), a structure of marvellous architecture and amazing biological activity. It belongs to the family of tetrapyrroles which includes inter alia the haems and the chlorophylls. This review begins with a brief overview of the biosynthesis of tetrapyrroles in general but then concentrates on recent research on B12 biosynthesis. The first main section reviews the biosynthesis of uro'gen III, the last common precursor of all natural tetrapyrroles, concentrating particularly on the three enzymes, porphobilinogen synthase, hydroxymethylbilane synthase and uro'gen III synthase. Crystal structures are available for the second of these enzymes and a new proposal is presented for its detailed mode of action. The second main section reviews the recent discovery of the complete biosynthetic pathway from uro'gen III to hydrogen- obyrinic acid in Pseudomonas denitrificans, which has revealed some beautiful and totally unexpected chemistry. A short section then describes the many similarities and some differences in the chemistry used by the micro-aerophilic organism Propionibacterium shermanii for the synthesis of coenzyme B12 compared with that seen in the aerobic Ps. denitrificans. Finally an account is given of the remarkable steps needed to complete the synthesis of the coenzyme in both organisms.
.
.
.
.
.
.
.
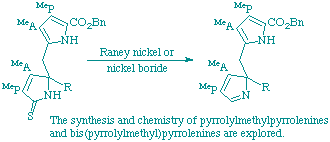 95
95
C. J. Hawker, W. M. Stark, A. C. Spivey, P. R. Raithby, F. J. Leeper and A. R. Battersby
"Biosynthesis of porphyrins and related macrocycles. Part 47. Synthesis and chemistry of 2H-pyrroles (pyrrolenines) related to the proposed spiro-intermediate for porphyrin biosynthesis"
J. Chem. Soc., Perkin Trans. 1,1998, 1493-1508.
Full Text
It is proposed that the biosynthesis of uroporphyrinogen III, the parent precursor of the natural porphyrins, chlorins and corrins, involves a pyrrolenine as a key intermediate, yet methods for the synthesis of such systems are not available. Novel routes for the synthesis of pyrrolenines by desulfurisation of unsaturated thiolactams have now been devised and the chemistry of such compounds has been explored. Enzymic experiments are carried out using a model pyrrolenine indicating that deletion of one of the pyrrole rings of the putative intermediate leads to loss of tight binding.
.
.
.
.
.
.
.
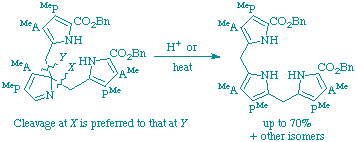 96
96
C. J. Hawker, A. C. Spivey, F. J. Leeper and A. R. Battersby
"Biosynthesis of porphyrins and related macrocycles. Part 48. The rearrangement of 2H-pyrroles (pyrrolenines) related to the proposed spiro-intermediate for porphyrin biosynthesis"
J. Chem. Soc., Perkin Trans. 1,1998, 1509-1518.
Full Text
It is proposed that the biosynthesis of uroporphyrinogen III, the parent precursor of the natural porphyrins, chlorins and corrins, involves a 2H-pyrrole (pyrrolenine) as a key intermediate. Model pyrrolenines have now been used to show that (a) pyrrolylmethylpyrrolenines readily undergo rearrangement in a way which matches that suggested in the biosynthetic proposal; (b) there is regioselectivity in the rearrangement which also parallels that required for the biosynthesis of uroporphyrinogen III. This work adds further support to the biosynthetic proposal by showing that all the required chemistry is both feasible and facile.
.
.
.
.
.
.
.
.
.
.
.
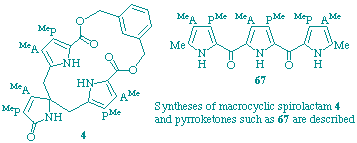 97
97
C. J. Hawker, P. M. Petersen, F. J. Leeper and A. R. Battersby
"Biosynthesis of porphyrins and related macrocycles. Part 49. Exploration of synthetic routes to analogues of the spiro-intermediate for porphyrin biosynthesis"
J. Chem. Soc., Perkin Trans. 1, 1998, 1519-1530.
Full Text
The proposed intermediacy of the spiro-pyrrolenine for the biosynthesis of uroporphyrinogen III has focussed attention on its synthesis. Several different approaches to close analogues of this compound are explored, including (a) the synthesis of a dilactone bridged dipyrrolic pyrrolenine, (b) deactivation of two of the pyrrole rings of the macrocycle by attaching 3-methoxycarbonyl groups and (c) approaches to spiro-macrocyclic compounds via dipyrroketones. The chemistry of the different types of synthetic intermediates is described.
.
.
.
.
.
.
.
.
.
.
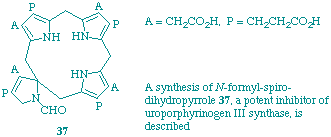 98
98
P. M. Petersen, C. J. Hawker, N. P. J. Stamford, F. J. Leeper and A. R. Battersby
"Biosynthesis of porphyrins and related macrocycles. Part 50. Synthesis of the N-formyl-dihydro analogue of the spiro-intermediate and its interaction with uroporphyrinogen III synthase"
J. Chem. Soc., Perkin Trans. 1, 1998, 1531-1539.
Full Text
The proposed intermediacy of the spiro-system for the biosynthesis of uroporphyrinogen III has focused attention on its synthesis. In this paper the approach that is explored is to carry a dihydropyrrole through the entire synthesis with the intention of converting it into a 2H-pyrrole (pyrrolenine) in one of the final steps. The chemistry of the different types of synthetic intermediates is described and also it is demonstrated that the N-formyl dihydropyrrole is a strong inhibitor of cosynthetase. The conclusion is reached that of all the possible routes to the spiro-pyrrolenine, that via the protected dihydropyrrole shows the greatest promise.
.
.
.
.
.
.
.
.
.
.
 99
99
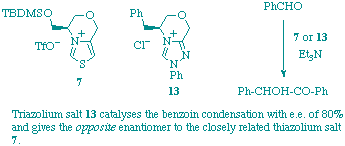
R. L. Knight and F. J. Leeper
"Comparison of chiral thiazolium and triazolium salts as asymmetric catalysts for the benzoin condensation"
J. Chem. Soc., Perkin Trans. 1,1998, 1891-1893.
Full Text
Chiral bicyclic 1,2,4-triazolium salts having a defined face of the heterocyclic ring hindered have been synthesised and they catalyse the benzoin condensation in good yield; the enantiomeric excesses obtained (up to 80%) are much better than with closely related thiazolium salts and the opposite enantiomer of benzoin predominates.
.
.
--- Next ten abstracts ----- Previous ten abstracts ---
.
.